Genome Sequencing
One very common thing done in genomics research is to sequence the DNA of an individual living thing. When all of the DNA in an individual is sequenced, that is called sequencing the whole genome. Too often, stories in the media describe this process in ways that are not accurate and sometimes over-hyped.
We want to help encourage better understanding of this key concept. This article is meant to clearly answer these questions:
- What is genome sequencing?
- What is it not?
- What can we learn from genome sequencing?
- What can we not learn from genome sequencing?
Complicated Legacies: The Human Genome at 20
A series of articles in the journal Science about the issues around human genome sequencing.
A series of articles in the journal Science about the issues around human genome sequencing.
Genome Sequencing
What is it and what is it good for?
You probably know of at least one living thing that has had its genome sequenced – if not, humans have, dogs and cats have, soybean and sunflower have. While the details of a newly sequenced genome will be published in an academic journal, any media coverage will typically focus is on how this new information revolutionises our understanding of the underlying biology. But what is genome sequencing and is the outcome always revolutionary?
Researchers use the term “genome” to describe all the DNA in an individual living thing and refer to the study of genomes as “genomics.”
Genome sequencing is now a widely used tool. Determining the DNA sequence allows us to identify all the genes – and other DNA types – that a genome contains. This can help us begin to understand how the genome and, ultimately the organism it comes from, works.
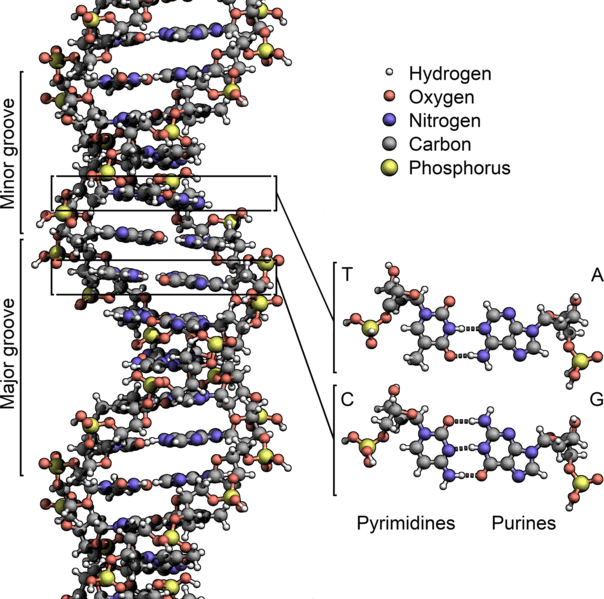
Sequencing a genome involves determining the order of the nucleotides – the As, Ts, Cs, and Gs that make up DNA – for all the DNA in an organism’s genome. With the right equipment and materials, sequencing sections containing several hundred or several thousand nucleotides is now a relatively straightforward process. However, sequencing entire genomes, especially those of plants and animals, is a more difficult task. This is due to both the size and complexity of these genomes.
The human genome, for example, was completed in 2003. The first human genome sequence took 13 years and US$ 3 billion – approximately a dollar for every nucleotide in the sequence. However, the technologies used to determine this first sequence have been replaced by “next-generation” approaches that have dramatically increased the speed and reduced the cost of DNA sequencing. We can now sequence a human genome in about a day for US$1000.
Next-generation sequencing methods work by breaking a genome into many smaller pieces. The samples being sequenced typically contain multiple copies of the genome and each is broken into a different collection of fragments. The pool of fragments is then sequenced and these sequences assembled into a genome sequence. How? Imagine you had a page from a book and cut it up line by line. If you didn’t know the story it would be difficult to put all the lines back in the correct order. But if you had other copies of the page and cut them up in different ways (e.g., from third word in a line to third word in next line) then by comparing the overlapping portions it would be much easier to reassemble the page.
There are two broad types of next-generation sequencing. “Short read” methods generate sequences that are 75-300 nucleotides long whereas those produced by “long read” methods can be 10,000 or more nucleotides in length. Each of these sequencing methods has pros and cons.
We can illustrate some of these using “repetitive DNA”. We can think of a genome as an encyclopedia with each entry corresponding to a segment of DNA. But imagine that every so often an entry in the encyclopedia was repeated over and over – in a genome this is called “repetitive DNA”. Short read sequencing can struggle with repetitive DNA because often the individual fragments will be too short to give us an unambiguous count of how many repeats there are. Long read sequencing overcomes this problem but currently has high rates of error and this can mean errors in the assembled sequence. Sequencing technologies are constantly improving but at present it is common to combine short and long read sequencing.
For small genomes, such as those of many bacteria, it is usually possible to obtain the complete sequence of the genome. But for plants and animals what we refer to as a “complete genome” often contains gaps. For example, about 4% of the human genome has yet to be sequenced, many of the gaps are thought to contain repetitive DNA.
Often the aim of genome sequencing is to identify what information a genome contains. What genes does it contain? Having a complete, or nearly so, catalogue of the genes for a genome and – if multiple genomes have been sequenced – how these genes differ between individuals is a major step forward. Indeed, this type of information is already making an impact. For example, genome sequencing is enabling personalized medicine, medical treatment that is tailored to an individual based on the gene variants in their genome.
However, the value of a genome sequence is easily overhyped and we need to be careful. Obtaining a genome sequence is a substantial achievement but it is not a magic bullet and alone a genome sequence can answer only a relatively narrow set of questions. And in many cases not the questions that are of greatest interest to researchers.
So why sequence genomes at all? While having a genome sequence may not answer a particular questions directly, having this data is often a necessary step towards the answers. Specifically, from a genome sequence we can infer a list of the genes contained within the genome and from that we can identify and then study in more detail genes or genetic pathways associated with particular characteristics.
The questions that researchers have often involve working out how the information in a genome is being used. The genome sequence tells us what genes are there but gives us no information about how or even whether a given gene is contributing to the growth, development and maintenance of the organism. The genome sequence is just the first step, it’s a big one but it is often just the start of the work that will be needed to address the questions researchers have.
So next time you read a media article about a new genome sequence remember that although we have taken a substantial step, we are likely some way off revolutionizing our understanding.
Researchers use the term “genome” to describe all the DNA in an individual living thing and refer to the study of genomes as “genomics.”
Genome sequencing is now a widely used tool. Determining the DNA sequence allows us to identify all the genes – and other DNA types – that a genome contains. This can help us begin to understand how the genome and, ultimately the organism it comes from, works.
Sequencing a genome involves determining the order of the nucleotides – the As, Ts, Cs, and Gs that make up DNA – for all the DNA in an organism’s genome. With the right equipment and materials, sequencing sections containing several hundred or several thousand nucleotides is now a relatively straightforward process. However, sequencing entire genomes, especially those of plants and animals, is a more difficult task. This is due to both the size and complexity of these genomes.
The human genome, for example, was completed in 2003. The first human genome sequence took 13 years and US$ 3 billion – approximately a dollar for every nucleotide in the sequence. However, the technologies used to determine this first sequence have been replaced by “next-generation” approaches that have dramatically increased the speed and reduced the cost of DNA sequencing. We can now sequence a human genome in about a day for US$1000.
Next-generation sequencing methods work by breaking a genome into many smaller pieces. The samples being sequenced typically contain multiple copies of the genome and each is broken into a different collection of fragments. The pool of fragments is then sequenced and these sequences assembled into a genome sequence. How? Imagine you had a page from a book and cut it up line by line. If you didn’t know the story it would be difficult to put all the lines back in the correct order. But if you had other copies of the page and cut them up in different ways (e.g., from third word in a line to third word in next line) then by comparing the overlapping portions it would be much easier to reassemble the page.
There are two broad types of next-generation sequencing. “Short read” methods generate sequences that are 75-300 nucleotides long whereas those produced by “long read” methods can be 10,000 or more nucleotides in length. Each of these sequencing methods has pros and cons.
We can illustrate some of these using “repetitive DNA”. We can think of a genome as an encyclopedia with each entry corresponding to a segment of DNA. But imagine that every so often an entry in the encyclopedia was repeated over and over – in a genome this is called “repetitive DNA”. Short read sequencing can struggle with repetitive DNA because often the individual fragments will be too short to give us an unambiguous count of how many repeats there are. Long read sequencing overcomes this problem but currently has high rates of error and this can mean errors in the assembled sequence. Sequencing technologies are constantly improving but at present it is common to combine short and long read sequencing.
For small genomes, such as those of many bacteria, it is usually possible to obtain the complete sequence of the genome. But for plants and animals what we refer to as a “complete genome” often contains gaps. For example, about 4% of the human genome has yet to be sequenced, many of the gaps are thought to contain repetitive DNA.
Often the aim of genome sequencing is to identify what information a genome contains. What genes does it contain? Having a complete, or nearly so, catalogue of the genes for a genome and – if multiple genomes have been sequenced – how these genes differ between individuals is a major step forward. Indeed, this type of information is already making an impact. For example, genome sequencing is enabling personalized medicine, medical treatment that is tailored to an individual based on the gene variants in their genome.
However, the value of a genome sequence is easily overhyped and we need to be careful. Obtaining a genome sequence is a substantial achievement but it is not a magic bullet and alone a genome sequence can answer only a relatively narrow set of questions. And in many cases not the questions that are of greatest interest to researchers.
So why sequence genomes at all? While having a genome sequence may not answer a particular questions directly, having this data is often a necessary step towards the answers. Specifically, from a genome sequence we can infer a list of the genes contained within the genome and from that we can identify and then study in more detail genes or genetic pathways associated with particular characteristics.
The questions that researchers have often involve working out how the information in a genome is being used. The genome sequence tells us what genes are there but gives us no information about how or even whether a given gene is contributing to the growth, development and maintenance of the organism. The genome sequence is just the first step, it’s a big one but it is often just the start of the work that will be needed to address the questions researchers have.
So next time you read a media article about a new genome sequence remember that although we have taken a substantial step, we are likely some way off revolutionizing our understanding.